AStream: An R package for annotating LC/MS metabolomic data

Package downloads:
- AStream: Windows binary (AStream_1.0.zip)
- AStream: Package source (AStream_1.0.tar.gz)
Package dependencies:
The AStream package depends on the "multtest", "Biobase" and “plotrix" packages. The following R commands install them from the CRAN and Bioconductor package repositories:
source("http://www.bioconductor.org/biocLite.R")
biocLite("Biobase")
biocLite("plotrix")
biocLite("multtest")
Package example data:
The AStream package includes an example set of 3,148 features and 20 samples that can be loaded using the following command:
data(astream.data,
package="AStream")
The intensities of this example data are also included in the following file: astream_data_example.xls
AStream tutorial
(b) Loading
user’s feature data
(a) data.norm
(b) isotope.search
(c) adduct.search
(d) printresults
The input data for the AStream R-package can
either be imported from an XCMS object or loaded as a data frame. The input data
must contain the basic information from a LC/MS metabolomics analysis, that is,
the mass to charge (m/z) value, the retention time and the detected intensities
for each feature in each of the samples analyzed.
a) Importing data from an XCMS object
XCMS (http://masspec.scripps.edu/xcms/xcms.php)
is one of the most commonly used open-source LC/MS preprocessing data software.
It performs noise filtering, peak selection and nonlinear time alignment when
multiple samples are analyzed. A complete review on this method and all the
used parameters can be obtained either from the website (http://metlin.scripps.edu/download/xcmsPreprocess.pdf)
or by typing vignette("xcmsPreprocess",
package = "xcms") into R.
In order to explain how an XCMS object can be
imported for an AStream analysis, we introduce here the commands required for a
typical XCMS analysis. First, you should place the R working directory on the
data path (when metabolomic samples are saved in different folders XCMS will
treat the samples of each folder as different groups). Once the work directory
has been set the typical XCMS analysis includes the following commands:
|
> library(xcms) |
|
> xset <- xcmsSet(snthresh = 4) |
|
> xset <- group(xset, minsamp = 3) |
|
> xset <- retcor(xset, family =
"s", plottype = "none", missing = 1, extra = 1) |
|
> xset <- group(xset, bw = 10,
minsamp = 3) |
|
> xset <- fillPeaks(xset) |
After running XCMS a LC/MS processed data
object (i.e. xset) is created. We can
now use AStream function import.xcms
to import the required data from this object:
|
> library(AStream) |
|
> input <- xcms.import(xset) |
The resulting data object for AStream is a list with two elements:
(a) A dataframe (input$data) with rows corresponding to the total number of features
(n=3,153 features in this case) and the first two columns indicating the m/z
values (first column) and retention times (second column). The remaining
columns contain the detected feature intensities for each individual, where the
column names correspond to the sample names (ID).
(b) A list (input$class) that contains the group/status of each sample.
b) Loading user’s feature data
Although XCMS
processing is highly recommended, users with their own feature dataset obtained
using any other analysis platform can also load their data in order to perform
an AStream data curation. In this
case is only necessary to format the data as an R list containing the two objects required by AStream as defined previously. An example of this type of input can
be directly loaded from the AStream
package. This example data consists of 20 LC/MS metabolomic samples randomly
divided into two groups (G1 and G2) with a total set of 3,148 features peaks:
|
> library(AStream) |
|
>
data(astream.data,package="AStream") |
|
> input$data[1:3,1:6] mzmed rtmed G1_1 G1_2 G1_3 G1_4 1 61.027944319865 426.307329158435 106306.455097724 127154.336715698 96099.2742362155 110844.540377937 2 64.0163388523736 389.533531615442 469463.71550916 462603.441696861 468016.930060338 470743.191450544 3 65.0190493567312 389.697081910654 19129.1374828996 17687.7633235852 16159.3797389168 16819.0124880034 |
|
> input$class[1:22,1] > input$class
class 1
G1 2
G1 3
G1 4
G1 5
G1 6
G1 7
G1 8
G1 9
G1 10
G1 11
G2 12
G2 13
G2 14
G2 15
G2 16
G2 17
G2 18
G2 19
G2 20
G2 |
Once AStream
data has been loaded to the defined workspace, we can start the analysis by
calling the first function data.norm
which performs outlier detection and feature selection based both on the
retention time differences and on the pairwise intensity correlation of all the
feature pairs. This function has five parameters:
·
std.outliers (default=3): This parameter is used to fix the
threshold value for the outlier detection method. Samples having a standardized
outlier score over this value will be excluded for further analysis. We can
also turn off outlier detection by setting std.outliers=0.
·
zero.missing (default=TRUE): Logical value, when TRUE intensity
values equal to zero are considered as missing values.
·
min.corr (default=0.75): Correlation threshold. Feature pairs
with a pairwise intensity correlation below this value will be excluded.
·
max.rt (default=3): Retention time difference threshold.
Feature pairs with a retention time difference over this value will be
excluded.
·
fplot (default=TRUE): Logical, if TRUE summary figures
are plotted.
The following two examples show how to perform
a default analysis and an analysis modifying several input. Relevant
information on the analysis is displayed on the R prompt such as the number of
samples excluded and the number of feature pairs selected:
|
EXAMPLE
1: Default analysis data.norm |
EXAMPLE
2: Modified parameters data.norm |
|
> feat1 <- data.norm(input) DATASET: -
#Features: 3148 -
#Samples: 20 - RT
limits: [ 1.6 , 1795.95 ] - M/Z
limits: [ 61.03 , 998.78 ] NORMALIZATION BY FEATURE MEDIAN... OUTLIER DETECTION... - Max
Dev allowed=3 -
SAMPLE #5 (G1_5) excluded: [3.929815,-2.897018] COMPUTING CORRELATION MATRIX... -
Zeros = Missing 0% 10%
20% 30% 40% 50% 60% 70% 80% 90% 100% FEATURE PAIRS SELECTION RESULTS: - MAX
RT DIFFERENCE ALLOWED: 3 - MIN
CORRELATION ALLOWED: 0.75 -
FEATURE PAIRS SELECTED:
11317 > names(feat1) [1] "data" "class" "mz" "rt" "feature.set"
"outliers"
"scores" |
> feat2 <- data.norm(input, max.rt =
2.5, min.corr = 0.60) DATASET: -
#Features: 3148 -
#Samples: 20 - RT
limits: [ 1.6 , 1795.95 ] - M/Z
limits: [ 61.03 , 998.78 ] NORMALIZATION BY FEATURE MEDIAN... OUTLIER DETECTION... - Max
Dev allowed=3 -
SAMPLE #5 (G1_5) excluded: [3.929815,-1.568823] COMPUTING CORRELATION MATRIX... -
Zeros = Missing 0% 10%
20% 30% 40% 50% 60% 70% 80% 90% 100% FEATURE PAIRS SELECTION RESULTS: - MAX RT DIFFERENCE ALLOWED: 2.5
- MIN
CORRELATION ALLOWED: 0.6 -
FEATURE PAIRS SELECTED: 18005 > names(feat2) [1] "data" "class" "mz" "rt" "feature.set"
"outliers"
"scores" |
|
EXAMPLE 1 SUMMARY PLOT |
|
|
|
|
|
EXAMPLE 2 SUMMARY PLOT |
|
|
|
|
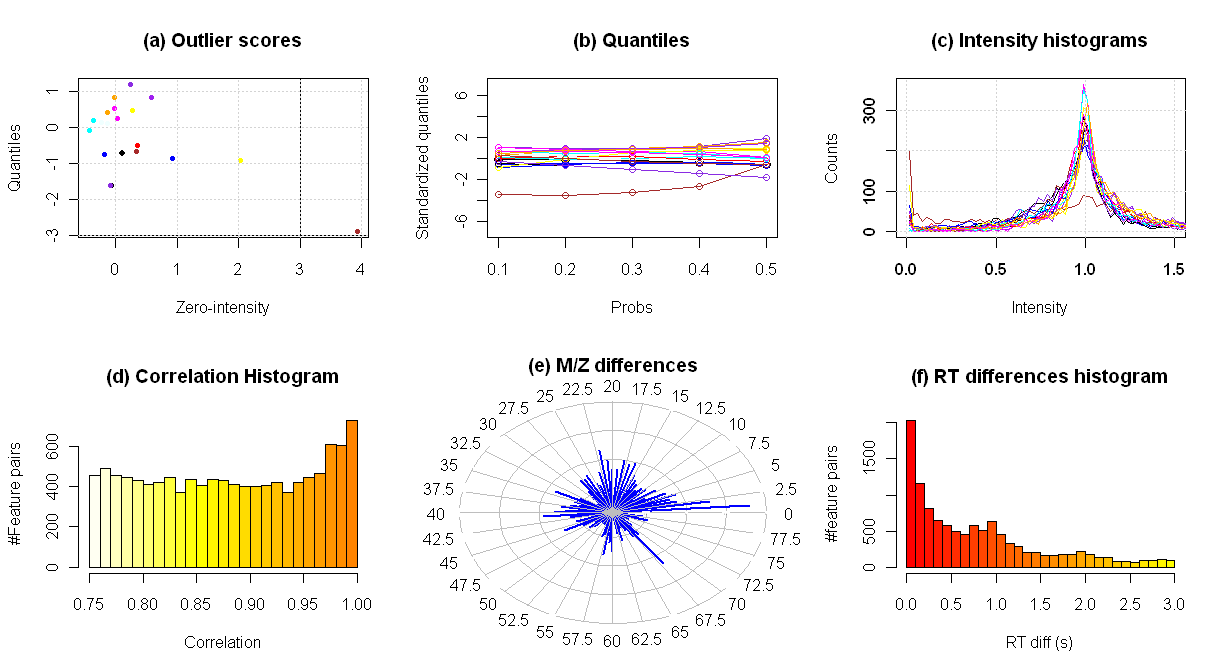

The plots that are generated can be very useful
to understand normalization and outlier exclusion process. Looking to the
following example, the plots show the following information:
(a) Outlier scores of each sample: The
scores computed from the number of features having zero intensity are
represented over the x-axis, while the scores computed from the intensity
quantiles are represented over the y-axis. In our example, one sample is
excluded because it zero-intensity score (x-axis) is greater than the default
score threshold std.outliers=3
(bottom-right orange point).
(b) Computed intensity quantiles (10%,
20%, 30%, 40% and 50%) for each sample. Each quantile has been standardized
using the values of all the samples.
(c) Normalized intensity distributions
of each sample. This figure also helps to detect the outlier sample (orange)
having a very high value at intensity 0 and a low value at intensity 1.
(d) Histogram of the pairwise feature
intensity correlation values that are greater than the specified threshold
(i.e. 0.75 in example 1 and 0.60 in example 2). We can observe the inflation
within the values near to 1 due to the existence of isotopes and adducts highly
correlated.
(e) Polar histogram of the m/z
differences within the selected feature pairs. The peaks due to 12C-13C
isotopes (m/zdiff=1.0033), [M+H]-[M+Na] adducts (m/zdiff=21.982)
and [M+H]-[M+NaHCOOH] adducts (m/zdiff=67.987) are clearly visible.
(f) Histogram of the retention time
differences that are greater than the specified threshold (i.e. 3.0 in example 1
and 2.5 in example 2). We can observe the inflation within the values near to 0
due to the existence of isotopes and adducts with almost the same retention
time values.
Once data.norm
function has been executed, the generated output list consists of all the
necessary data for the isotope and adduct search analysis:
·
data: Numeric matrix with the intensities of all the
features (rows) from all samples (columns). In the present example, there are n=19
final samples after excluding one sample having an outlying signal profile.
> dim(feat1$data)
[1]
3148 19
·
class: Group label of each included sample.
> feat1$class[,1]
[1]
"G1" "G1" "G1" "G1" "G1"
"G1" "G1" "G1" "G1" "G1"
"G2" "G2" "G2" "G2" "G2"
"G2" "G2" "G2" "G2" "G2"
·
rt and mz:
Retention time and m/z values of each feature.
·
feature.set: Set of feature pairs that will be
analyzed in order to find isotopes, adducts or fragments. This set contains all
the pairs that have passed the correlation and retention time filter thresholds
specified in data.norm.
> feat1$feature.set[1:10,]
Index feature 1 Index feature 2 Correlation %samples
RTdiff M/Zdiff
[1,]
1 35 0.7722203 100
1.16187443 23.019208
[2,]
1 153 0.7765506 100
0.96656101 60.024970
[3,]
2 97 0.8418388 100
0.25841026 41.026011
[4,] 2 195 0.8360164 100
1.84080403 67.986864
[5,]
2 542 0.7992878 100
0.91842920 135.981157
[6,]
4 5 0.8114268 100
0.01603013 0.988777
[7,]
4 11 0.9074511 100
0.00960247 6.509211
[8,]
4 14 0.8939059 100
0.02002257 7.508971
[9,]
4 113 0.7943203 100
0.18891644 40.972375
[10,] 4 115 0.7504487 100
0.20864333 42.987411
·
outliers: Indexes of the outlier excluded
samples within the initial sample group.
·
scores: Outlier scores of each sample within the
initial sample group. Two scores are provided, the first corresponding to the
number of features with zero intensity and, the second, corresponding to the
intensity quantiles. In the present example, the scores corresponding to the
outlier sample (sample #5) have been highlighted. The outlying values from this
sample are indicative of a potential problem either in the sample collection
phase or during the LC/MS data acquisition process:
> feat1$scores
$int_0
[1] 0.11228042
-0.17964868 0.26947301 -0.13473651 3.92981477 0.24701693
-0.42666560 -0.02245608 -0.02245608
[10] 0.58385819
-0.06736825 0.35929735 0.92069946
2.02104760 -0.22456084 0.33684127
-0.08982434 -0.35929735
[19] 0.02245608
-0.13473651
$int_1
[1] -0.7257414 -0.7654592 0.4761860
0.1038420 -2.8970182 1.1807430 -0.1038420 0.5233418
0.8306186 0.8248309
[11] -1.6179217 -0.5120058 -0.8800208 -0.9313024 0.1438316 -0.6664879 -1.6177946 0.1941364
0.2443339 0.4189088
The following step in the analysis is to search
for isotopes within the feature pairs selected by data.norm. The function isotope.search
performs this search by grouping all those features that fulfill the
expected m/z differences between carbon isotopes of the same molecular
compound:
·
1st
13C-isotope peak difference: m/z[13M]
- m/z[12M] = 1.0033
·
2nd
13C isotope peak difference: 2*(m/z[13M]
- m/z[12M]) = 2.0066
isotope.search function has three input parameters:
·
ftlist: Dataset list as returned by data.norm.
·
mz.tol (default=3e-3): Tolerance for m/z values.
||m/z[featurei] - m/z[featurej]|
- 1.0033| < mz.tol à features i and j will be grouped as 12C-monoisotopic
peak and first 13C-isotope peak of the same molecular compound
||m/z[featurei] - m/z[featurej]|
- 2.0066| < mz.tol à features i and j will be grouped as 12C-monoisotopic
peak and second 13C-isotope peak of the same molecular compound
·
fplot (default=TRUE): Logical, if TRUE summary figures
are plotted.
The output of this function is the same than
the input F but with an added “isotopes” component which contains the
obtained information relative to isotopes. In the present example:
|
EXAMPLE
1: isotope.search |
|
> feat1 <- isotope.search(feat1,
mz.tol = 3e-3) PE ANALYSIS:
- M/Z tolerance: 0.003u - C12-isotope m/z: 12u - C13#1-isotope m/z: 13.00335u -
C13#2-isotope m/z: 14.0067u -
13C#1/12C-isotope: 378 feature pairs - 13C#2/12C-isotope: 71
feature pairs -
Total number: 449 isotope patterns > feat1$isotopes[1:10,]
Index-12C Index-13C1 Index-13C2 Intensity-12C Intensity-13C1
Intensity-13C2 [1,] 12 16 0 3090537.8 130994.89 0.00 [2,] 13 17 0 1079753.1 44512.43 0.00 [3,] 23 25 0 175807.6 25626.86 0.00 [4,] 39 46 0 272485.3 19822.19 0.00 [5,] 41 47 0 654658.6 41185.92 0.00 [6,] 42 48 0 1151918.4 66914.15 0.00 [7,] 45 52 0 460785.7 43766.34 0.00 [8,] 86 88 0 514900.2 10721.60 0.00 [9,] 98 102 0 1796847.8 85156.37 0.00 [10,]
98 0 108 1796847.8 0.00 26828.69 |
|
EXAMPLE 1 SUMMARY PLOT |
|
|

In the present example, feat1$isotopes contains the isotope grouping results with the
feature indexes linked to the obtained isotope patterns. The summary plot shows
the mean intensity ratios between first and second 13C-isotopic
peaks and 12C–monoisotopic peak. Given the lower abundance of C13 species,
the detected patterns must have intensity ratios lower than 1 as the abundances
(intensities) of 13C isotopes are further lower than 12C
isotopes.
The
adduct.search function performs the last data reduction step of the AStream
analysis. The input parameters are:
·
ftlist: Dataset list as returned by isotope.search.
·
mz.tol (default=3e-3): Tolerance for m/z differences.
·
adducts (default=0): According to the particular
technical parameters of the LC/MS acquisition process, the user can define its
own set of adducts to perform the adduct search step. This must be a data.frame
object with two columns, the first indicating the adduct mass and the second
the corresponding label. The protonated compound (i.e. [M+H]), is the reference
adduct and therefore must always be present in this data frame. By default, if
no data.frame object is used, a predefined set of adducts will be used (see
below).
·
onlyIsotopes (default=TRUE): If True, the adduct search will
only take into account those features that have been previously associated to
an isotope pattern. This option is highly recommended for robustness in
metabolite annotation.
·
fplot (default=TRUE): Logical, if TRUE summary figures are
plotted.
Two examples are showed below, the first one
with the default set of adducts and the second using an alternative set of
adducts loaded using the “adducts” argument.
|
EXAMPLE
1: adduct.search |
|
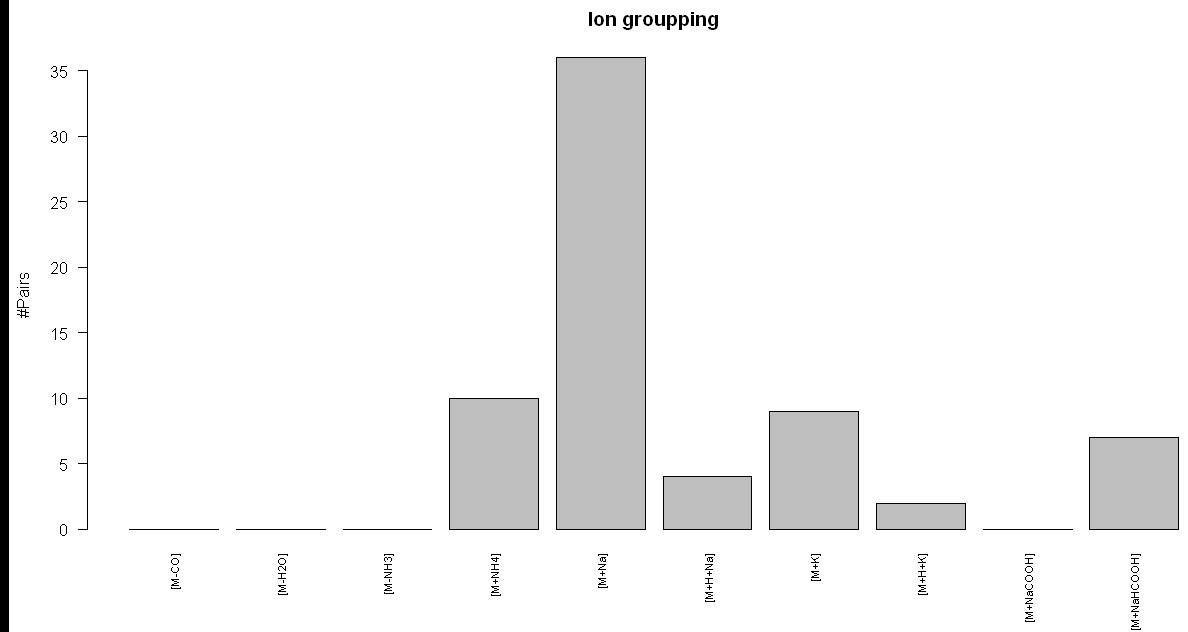
> res <- adduct.search(feat1) ADDUCT ANALYSIS: - M/Z
tolerance: +/-0.003u - 0
[M-CO]adducts. - 0
[M-H2O]adducts. - 0
[M-NH3]adducts. - 10
[M+NH4]adducts. - 36
[M+Na]adducts. - 4 [M+H+Na]adducts. - 9
[M+K]adducts. - 2
[M+H+K]adducts. - 0
[M+NaCOOH]adducts. - 7
[M+NaHCOOH]adducts.
|
|
EXAMPLE 2: adduct.search |
|
> adductlist <- data.frame(cbind(c(1.007276,
18.033823,22.989218,38.963158,68.99472), +
c("[M+H]","[M+NH4]","[M+Na]","[M+K]","[M+NaHCOOH]"))) > adductlist
X1 X2 1 1.007276 [M+H] 2
18.033823 [M+NH4] 3
22.989218 [M+Na] 4
38.963158 [M+K] 5
68.99472 [M+NaHCOOH] > res2 <- adduct.search(feat1,
adducts=adductlist) ADDUCT ANALYSIS: - M/Z
tolerance: +/-0.003u - 10
[M+NH4]adducts. - 36
[M+Na]adducts. - 9
[M+K]adducts. - 7
[M+NaHCOOH]adducts. > res2[1:20,]
C12isotope C13#1 isotope C13#1
ratio C13#2 isotope C13#2ratio [M+NH4] [M+Na] [M+K] [M+NaHCOOH] [1,] 12 16 0.04238579 0 0.00000000 0
0 0 0 [2,] 13 17 0.04122463 0 0.00000000 0
0 0 0 [3,] 23 25 0.14576649 0 0.00000000 0
0 0 0 [4,] 39 46 0.07274591 0 0.00000000 0
0 0 0 [5,] 41 47 0.06291206 0 0.00000000 0
0 0 0 [6,] 42 48 0.05808931 0 0.00000000 0
0 0 0 [7,] 45 52 0.09498200 0
0.00000000 0
0 0 0 [8,] 86 88 0.02082268 0 0.00000000 0
0 0 0 [9,] 98 102 0.04739209 108 0.01493098 0
0 0 0 [10,]
99 103
0.04784580 0 0.00000000 0
171 0 0 [11,]
102 108
0.31505204 0 0.00000000 0
0 0 0 [12,]
115 117 0.18524925 0 0.00000000 0
0 0 0 [13,]
138 140 0.05027759 0
0.00000000 0
0 0 0 [14,]
144 148 0.05115318 0
0.00000000 0
0 0 0 [15,]
145 149 0.05692672 0
0.00000000 0
0 0 0 [16,]
146 150 0.05210245 0
0.00000000 0
0 0 0 [17,]
158 161 0.04571414 163
0.01947436
246 261 339 0 [18,]
161 163 0.42600300 0 0.00000000 0
0 0 0 [19,]
184 192 0.06698323 0
0.00000000 0
0 0 0 [20,]
185 193 0.06751841 0
0.00000000 0
292 0 0 |

The output of this function is a list of
feature groups that can be definitively related to a single compound. For each
compound (row) the indexes of the related features corresponding to isotopes
and adducts are given in the successive columns. If we want to visualize all
the features that have been related to a specific compound we can call the
function plotcompound using the
compound index in the adduct.search
output (i.e. res). Here we show the
distribution of two different detected compounds (#10, #71) obtained from the
previous analysis:
|
EXAMPLE: plotcompound |
|
> res[10,] C12isotope C13#1 isotope C13#1 ratio C13#2 isotope C13#2ratio [M-CO] [M-H2O] [M-NH3] [M+NH4] [M+Na]
[M+H+Na] 99 103 0.0478458 0 0.0 0 0 0
0 171 0 > plotcompound(10, feat1, res)
> res[71,] C12isotope C13#1 isotope C13#1 ratio C13#2 isotope C13#2ratio [M-CO] [M-H2O] [M-NH3] [M+NH4]
[M+Na] [M+H+Na] 733 741 0.1313409 0 0.0
0 0 0 831 856
0 > plotcompound(71, feat1, res)
|


After applying AStream’s analysis flow, the non-redundant and robust set of
metabolic features can be exported as a tab delimited text file using the printresults function. This table
summarizes the principal characteristics of each compound an provides a direct
link to the METLIN metabolomics database (http://metlin.scripps.edu/metabo_search.php).The input parameters for this
function are:
·
filename: name of the file where the results
will be extracted.
·
ftlist: Output returned by isotope.search.
·
results: Output returned by adduct.search.
·
range (default = 1e-2): m/z tolerance for the search in the
Metlin database.
·
Mzsort (default = FALSE ): If TRUE results will be sorted by m/z
value. If FALSE, results will be sorted by P-Value if there are two groups of
samples.
In the present example, the output file can be
generated as it follows:
|
EXAMPLE: printresults |
|
> printresults("results.txt",
feat1, res) |
The tab delimited text file where the results
are extracted using the printresults
function has the following fields. Example values are referred to the example
line below:
·
IndexFeature: Index within the original feature
set (AStream input) of the feature
labeled as [M+H]+ (i.e. 486).
·
MassSubmitted: Expected m/z value of the compound
[M] ([M+H]+ - [H]) (i.e. 188.1045).
·
M/Z: m/z value of the feature labeled as [M+H]+
(i.e. 189.1121).
·
RT: retention time value of the feature labeled as
[M+H]+ (i.e. 1050.36).
·
P-Value: If two groups of samples have been
provided, P-Value of the intensity association test between those groups (i.e. 0.00625).
·
FoldChange: If two groups of samples have been
provided, intensity fold change between those groups (i.e. 3.57).
·
N1: If two groups of samples have been provided,
number of samples of the first group having non zero intensity at the feature
labeled as [M+H]+ (i.e. 9).
·
N2: If two groups of samples have been provided,
number of samples of the second group having non zero intensity at the feature
labeled as [M+H]+ (i.e. 10).
·
Metlin: Link to the Metlin database (i.e. http://metlin.scripps.edu/metabo_list.php?mass_min=188.0945&mass_max=188.1145).
·
13C#1-isotope: If the 13C-isotope feature has been identified
this field contains 4 numbers separated by a space corresponding to the index
of the 13C-isotope within the original feature set, the intensity ratio between
13C-isotope and 12C-isotope features, the pairwise intensity correlation of
those features and their retention time difference (i.e. 495 0.1
1 0.208).
·
13C#2-isotope: If the 14C-isotope feature has been
identified this field contains 4 numbers separated by a space corresponding to
the index of the 14C-isotope within the original feature set, the intensity
ratio between 14C-isotope and 14C-isotope features, the pairwise intensity
correlation of those features and their retention time difference (i.e. NA).
·
Adducts: If there are one or more adduct features
identified, they are listed here giving their label and their index within the
initial feature set (i.e. [M+NH4]:573 [M+Na]:597).
·
NFG: Number of features grouped within this
compound (i.e. 4).
Example line in the present example:
|
IndexFeature |
MassSubmitted |
M/Z |
RT |
P-value |
FoldChange |
N1 |
N2 |
Metlin |
13C-isotope |
14C-isotope |
Adducts |
NFG |
|
486 |
188.1045 |
189.1121 |
1050.36 |
0.00625 |
3.57 |
9 |
10 |
http://metlin.scripps.edu/metabo_list.php?mass_min=188.0945&mass_max=188.1145 |
495
0.1 1 0.208 |
NA |
[M+NH4]:573
[M+Na]:597 |
4 |
|
MS/MS |
||||
|
|
|
|
|
